

What are the causes of insulin resistance?
The accumulation of substrate
Simply put, years of excess intake of high glycaemic load (GL) carbohydrates and saturated fats found in the Western diet can overwhelm the biochemical pathways of energy production in the body. After a period of time the body is not able to keep up with the demand of processing all the energy molecules and energy is stored as fat and glycogen. At some point the cell is overwhelmed with excess energy molecules (end‑products and intermediary substrates) and will not allow further sugar or saturated fatty acid uptake.
One of the ways in which the body blocks energy entrance is through down regulation of GLUT 4 transport within the muscle cell; therefore, glucose cannot be transported into the cells. In the early stages of IR, glucose is stored as glycerol (along with the excess fatty acids) in the fat cells rather than being metabolised and used for energy.
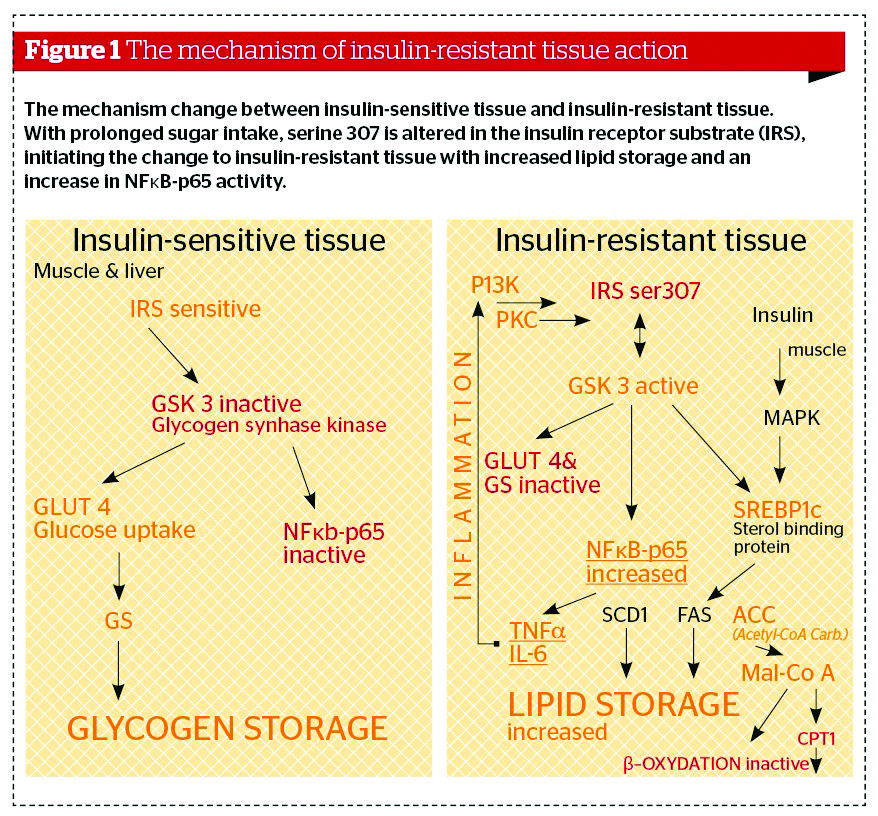
If excess energy intake continues, the fat cells become saturated with energy and glucose, as well as the fatty acids being blocked from entering the fat cells. The circulating glucose is converted to lipids by the liver. This is a predominant pathway to increased triglyceride levels in insulin-resistant conditions. Furthermore, overconsumption of calories with a high GL, and particularly the metabolism of lipids, leads to metabolic products that result in glycosylation and phosphorylation of the insulin receptor substrate (IRS) (Figure 1) on serine residues (serine 307), terminating the insulin signal.
Counter regulatory hormones
over-powering insulin
Insulin’s natural activity is to store glucose, while the counter regulatory hormones (CRHs) (cortisol, human growth hormone (HGH), glucagon, and catecholamines) try to prevent fuel storage in order to keep glucose going to the brain. They attempt this through increased gluconeogenesis, glycogenolysis, and increased lipolysis, as well as limited glucose use.
The escalating levels of CRHs start early in IR, as the counter regulatory hormones are the first to be released under stressful conditions. The more stresses that occur on a daily basis, such as nutritional, emotional, chemical, physical or hormonal, and the longer these stresses are present, will stimulate chronic release of higher levels of the CRHs. To maintain homeostasis of blood glucose levels in a chronic stressful environment (which is our everyday life), requires a shift in both the baseline and postprandial insulin levels. A point is reached at which the system starts to decompensate. The pancreas cannot keep up production and secretion of insulin to counter the effects of the CRHs, and the metabolic derangement of metabolic syndrome becomes apparent as the four counter regulatory hormones over-power the insulin. Stress, primarily through hyperactivation of the hypothalamic-pituitary-adrenal (HPA) axis, appears to contribute to the accumulation of visceral fat tissue and vice versa6. Therefore, cortisol is considered to be a silent killer in modern society. Recent data suggests that a number of neuroendocrine and inflammatory mechanisms are mobilised during chronic stress in the development of central obesity and metabolic syndrome7. Obesity itself seems to constitute a chronic stressful state and may cause HPA axis dysfunction. Obesity may be considered a systemic, low-grade, inflammatory condition with increased CRHs7.
Genetic propensity
There has been more awareness with regard to the fact that IR is becoming more apparent at a younger age and it is no long a problem exclusively related to the older population.
Sullivan presents one possible explanation: ‘It is theorised that the metabolic syndrome may be a manifestation of the profound mismatch between our present environment and previous circumstances that have molded evolutionary selection’8.
Familiar transmissions and variations in different ethnic distributions suggest that IR is genetically predisposed. It is likely that impaired insulin action has been the result of a number of inherited mutations in a variety of genes each with minor effects. Baroni et al support this view: ‘Several studies indicate the possible role of mutations of the insulin receptor substrate-1 (IRS-1) gene in the pathogenesis of insulin-resistance and suggest a possible interaction between the IRS-1 gene and obesity, either by an effect on the development of obesity or by causing or aggravating the obesity-associated with insulin resistance’9.
Recent data suggest that marked impairment in insulin signalling is happening in adipose tissue long before glucose intolerance in individuals with genetic predisposition for type 2 diabetes10.
Persistent organic pollutants

A popular statement is that our genes have not changed, but our environment has and that is what creates an epidemic. It has been found that a strong dose–response relation exists between serum concentrations of six persistent organic pollutants and the prevalence of type 2 diabetes11. Persistant organic pollutants (POPs) have been associated with type 2 diabetes risk by increasing IR; POPs may interact with obesity to increase the risk of type 2 diabetes11. It has been postulated that some common dietary ingredients like mono-oleoyl glycerol, iron, and saccharin are capable of producing long term hyperinsulinaemia owing to their toxic activity on beta cells, which react with the hyper-production of insulin12. Similar activity was found from arsenic, leading to an increased mortality rate from cardiovascular disease and diabetes13. A recently published article stated that Bisphenol A (BPA), in connection with insulin, can accelerate the conversion of 3T3-L1 fibroblasts into adipocytes14. There is certainly enough scientific data to explain why IR is currently a global epidemic problem.
Inflammation (the link between abdominal obesity and IR)
Research has implicated dysregulated inflammatory processes in the development of IR and cardiometabolic syndrome. Some inflammation-induced cytokines have been shown to be expressed during IR and metabolic syndrome, and mainly produced by visceral fat (Figure 2)15. For example, cytokines such as IL-1β, TNF-α, and interferon-gamma (IFN-γ) have been shown to play a role in cytokine-induced beta cell dysfunction in diabetes, as long as they are under nuclear factor-κB (NF‑κB) control. Modalities that regulate NF-κB expression may be beneficial in reducing IR and new medical research is focusing more in that direction16-18. The majority of these inflammatory cytokines are produced in metabolically-active visceral fat. In fact, when visceral fat was extracted from aged rats accounting for approximately 18% of their total body fat, it decreased the expression of TNF-α and a delayed onset of diabetes was achieved19. New functional medicine approaches of cardiometabolic syndrome include necessary treatment of the underlying inflammation (Figure 3).
Inflammation consequences mediated via inflammatory cytokines leading to increased cardiometabolic risks are:
- Endothelial dysfunction
- Disruption of nitric oxide (NO) synthesis
- Arterial plaque deposition
- Oxidative stress
- Mitochondrial suppression
- Islet cell abnormalities
- Reduced glucose tolerance.
Vitamin deficiency
A number of scientific studies20-22 have proven that low vitamin D levels are associated with IR, and that vitamin D supplementation improved IR. There is an inverse correlation between 25-hydroxy vitamin D, metabolic syndrome, and diabetes. Optimal levels of vitamin D are essential for normal insulin release, glucose tolerance, and for the regulation of serum calcium and free fatty acid metabolism. Low vitamin D levels have been implicated in heart failure, myocardial dysfunction, and sudden cardiac death. The insulin receptor gene promoter has been found to be sensitive to vitamin D supplementation. These effects were accompanied by a normalisation of the number of insulin receptors without altering receptor affinity, but improving the insulin response to glucose transport in adipocytes20-22. Checking vitamin D levels is a necessary part of extensive work towards diagnosing IR.
5-Hydroxytryptophan and serotonin connection with insulin resistance
A number of human studies with 5-hydroxytryptophan (5-HTP)23-25, the precursor of serotonin, have found to be beneficial in treating IR. A well-known mechanism for developing IR from overwhelming metabolic pathways with high carbohydrate diet can be counteracted with a low GL carbohydrate. The problem is that some individuals compulsively snack on carbohydrates with a significant GL, which increases tryptophan/serotonin in the brain, temporarily reducing anxiety and depression in these individuals. This also increases the release of insulin to a degree that eventually becomes pathological. Providing an alternative, non-insulin-driven way to increase brain serotonin with 5-HTP supplements may help reduce IR, especially by reducing carbohydrate intake and decreasing hyperinsulinaemia23. Some studies identified that serotonin receptor (5-HT) are a regulator of insulin secretion, and thereby contribute a function to the co-localisation of serotonin and insulin in pancreatic β-cells through the independent receptor signalling mechanism24.
There are a number of mechanisms in which abnormal liver function in metabolic syndrome can cause disturbances in brain activity and mood. The liver enzyme tryptophan oxygenase is activated by gluconeogenesis and it converts tryptophan to kynurenine, which shifts away from making serotonin and into the pathway for making glucose. Replenishing serotonin with 5-HTP can stabilise mood in this condition25.

